AlphaFold 2: A Breakthrough in Protein Structure Prediction and the Role of Cloud Computing
Introduction
Predicting protein structure has long been one of biology’s grand challenges. The three-dimensional structure of proteins determines their function and behavior, yet deducing this structure from an amino acid sequence alone remained unsolved for decades. Experimental methods such as X-ray crystallography require immense time and resources, making computational solutions highly desirable. However, the complexity of protein folding and the limited availability of experimental data posed significant challenges.
The introduction of AlphaFold 2 by DeepMind in 2020 marked a transformative moment in both artificial intelligence and structural biology. The model’s unprecedented accuracy in predicting protein structures culminated in the awarding of the 2024 Nobel Prize in Chemistry to the researchers behind it. This blog post examines the innovations behind AlphaFold 2, its impact on biological research, and its broader implications for artificial intelligence, life sciences, and cloud computing.

Cloud Computing in Biomedicine
Advancements in artificial intelligence and deep learning have driven a significant shift in how biomedical research is conducted. Cloud computing plays a crucial role in enabling large-scale computations, making high-performance computing accessible to researchers worldwide.
Key Applications of Cloud Computing in Biomedicine:
- Scalability: Cloud platforms provide on-demand access to vast computational resources, essential for running complex AI models like AlphaFold.
- Data Storage & Accessibility: Biological datasets, including genomic sequences and protein structures, require extensive storage. Cloud-based solutions ensure secure and efficient access to this data.
- Collaboration & Democratization: Cloud technologies allow researchers across institutions to collaborate in real time, reducing the barrier to entry for developing and using advanced AI tools.
- Accelerated Drug Discovery: AI-driven predictions, supported by cloud computing, enable faster identification of drug targets and protein interactions, expediting pharmaceutical research.
Cloud computing has been instrumental in supporting projects like AlphaFold, enabling the model to perform large-scale computations that would otherwise be infeasible on standard hardware.
Context and State of the Art Before AlphaFold 2
Before AlphaFold 2, protein structure prediction relied on two main approaches:
- Physics-Based Simulations: Tools like Rosetta and AMBER attempted to model molecular interactions through thermodynamic simulations. However, the immense computational complexity made these methods impractical for larger proteins.
- Evolutionary Analysis: Methods like PSICOV and I-TASSER used evolutionary information from the Protein Data Bank (PDB) and genomic sequencing to predict structures. While beneficial, they fell short in accuracy for proteins lacking known structural homologs.
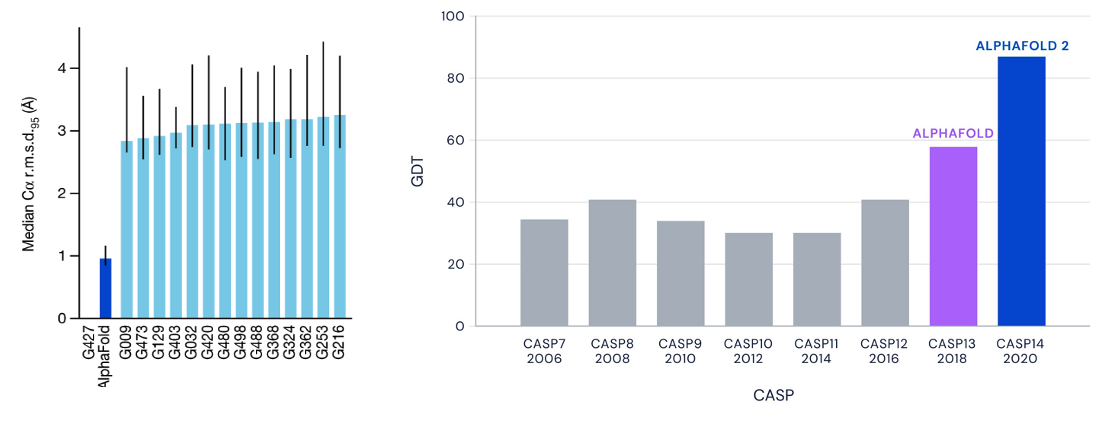
The Critical Assessment of Protein Structure Prediction (CASP) competition tracked progress in this field. Before AlphaFold 2, even the best models often had deviations exceeding 3-4 Ångstroms from experimental structures, making reliable predictions difficult (images below). While deep learning started showing promise, challenges remained in handling the vast number of conformations a protein could adopt and modeling long-range amino acid interactions effectively.
Breaking Down AlphaFold 2
AlphaFold 2’s architecture consists of three main modules:
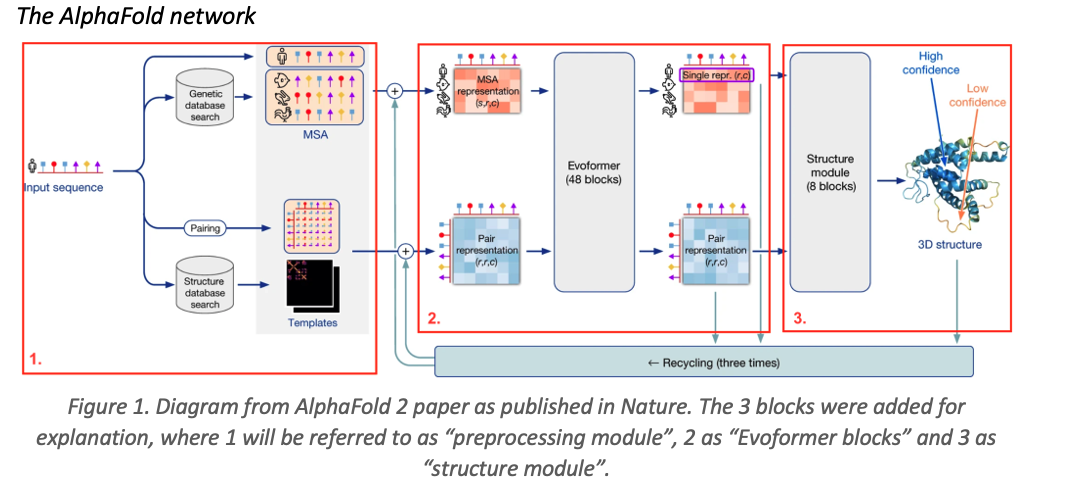
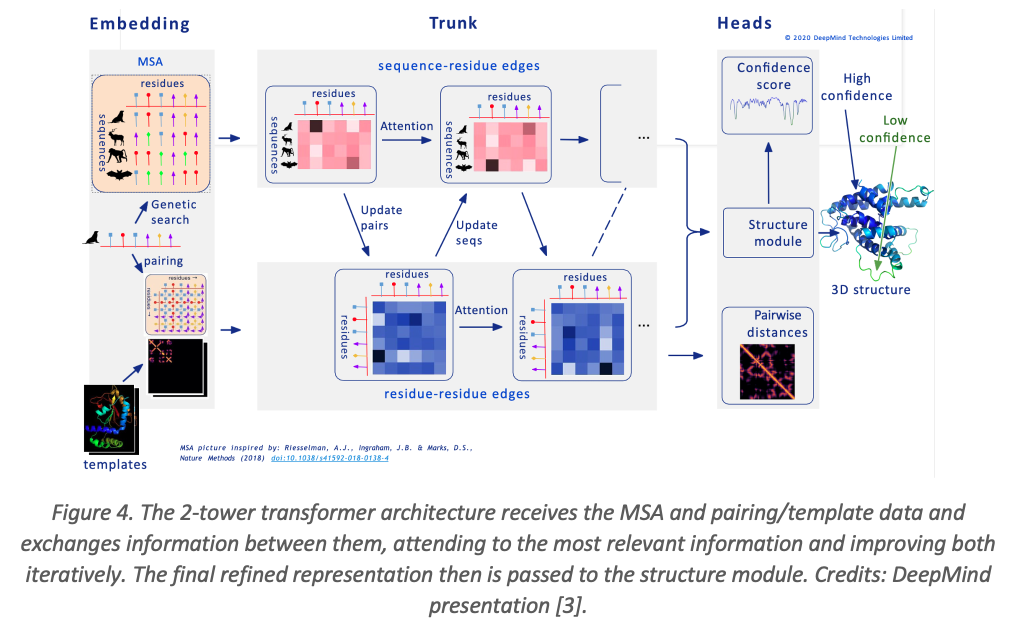
1. Preprocessing Module
The model processes the input amino acid sequence using three data representations:
- Multiple Sequence Alignment (MSA): Identifies homologous proteins, helping detect evolutionary correlations.
- Pair Representation: A matrix that captures amino acid interactions.
- Templates: Uses known protein structures to guide predictions.
2. Evoformer Blocks
The Evoformer module utilizes a dual-transformer architecture with separate processing for MSA and pairwise interactions. It refines structural predictions iteratively using:
- Efficient Attention Mechanisms: Reduces memory cost while maintaining accuracy.
- Triangular Attention Mechanism: Ensures distance constraints between amino acids.
- 48 Iterations: Progressive refinement for improved accuracy.
3. Structure Module
This module converts the learned representations into 3D atomic coordinates while maintaining physical constraints. The Invariant Point Attention (IPA) mechanism ensures that predictions remain accurate regardless of spatial orientation.
Additionally, AlphaFold 2 incorporates:
- pLDDT Confidence Measure: Estimates prediction reliability per amino acid.
- Self-Distillation Training: Expands training data by predicting structures of uncharacterized proteins and incorporating high-confidence predictions into model retraining.
The Role of Cloud Computing in AlphaFold 2
AlphaFold 2’s success was heavily reliant on cloud-based infrastructure and high-performance computing. Training and running deep learning models of this scale require massive amounts of computational power, which would be infeasible on traditional academic resources.
How Cloud Computing Enabled AlphaFold 2:
- Massive Computational Power: AlphaFold 2 was trained using 128 TPUv3 cores, equivalent to hundreds of high-end GPUs, made possible through cloud-based platforms.
- Data Storage & Processing: Handling the vast amounts of protein sequence data required efficient cloud storage and distributed computing to optimize processing speed.
- Open Access & Democratization: DeepMind’s decision to make AlphaFold’s predictions available via cloud-hosted databases has allowed researchers worldwide to access protein structures without requiring their own computational infrastructure.
- AI-Powered Drug Discovery Pipelines: Pharmaceutical companies now leverage cloud-based AlphaFold models to identify and develop new drugs more efficiently.
Strengths and Limitations
Strengths
- Near-Experimental Accuracy: At CASP14, AlphaFold 2 achieved median backbone accuracy of 0.96 Å RMSD for template-available domains and 1.71 Å for challenging targets.
- End-to-End Learning: Unlike previous approaches that relied on multiple separate steps, AlphaFold 2 integrates everything into a single trainable architecture.
- Cloud-Based Accessibility: AlphaFold 2’s cloud-hosted structure predictions make protein modeling widely available to researchers.
Limitations
- Dependence on MSA: The model struggles with proteins that lack homologous sequences, such as designed proteins and antibodies.
- Static Predictions: AlphaFold 2 does not capture conformational dynamics, crucial for understanding protein function.
- High Computational Demand: Training requires significant cloud-based infrastructure, limiting accessibility for offline use.
Future Directions and Alternative Approaches
While AlphaFold 2 revolutionized protein structure prediction, its limitations have led to the development of complementary models:
- AlphaFold 3 (2024): Extends predictions to protein-ligand, nucleic acid, and small molecule interactions.
- RoseTTAFold: A simpler alternative requiring fewer resources.
- Chai-1 Model: Open-source model for protein-ligand interactions, rivaling AlphaFold 3.
- ProteinMPNN & RFdiffusion: Flip the paradigm by generating amino acid sequences from desired 3D structures, aiding in de novo protein design.
- Meta AI’s ESM3: Uses deep learning to simulate protein evolution and generate novel protein sequences.
Conclusion
AlphaFold 2 represents a groundbreaking moment in artificial intelligence and life sciences, proving the power of deep learning in solving complex biological problems. Its accuracy approaches experimental methods, significantly impacting drug discovery, protein design, and fundamental biology.
Moreover, cloud computing has been a fundamental enabler of AlphaFold 2’s success, providing the computational infrastructure necessary to train, store, and deploy large-scale AI models. As cloud-based AI models continue to evolve, their applications in biomedicine will only expand, leading to faster discoveries and broader accessibility.
The awarding of the 2024 Nobel Prize in Chemistry to AlphaFold’s creators further underscores the model’s transformative role in scientific research, marking a new era in AI-driven discovery.
References
- Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
- 2024 Nobel Prize lectures in chemistry - David Baker, Demis Hassabis and John Jumper
- Guest lecture on the “Geometric Deep Learning” course taught in the African Master in Machine Intelligence in July 2022 by Russ Bates (DeepMind). AIMMI Seminar: AlphaFold - Russ Bates Presentation.
- Oxford Protein Informatics Group blog - AlphaFold 2 is here: what’s behind the structure prediction miracle - Oxford Protein Informatics Group
- Alford RF, et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput. 2017 Jun 13;13(6):3031-3048. doi: 10.1021/acs.jctc.7b00125. Epub 2017 May 12. Erratum in: J Chem Theory Comput. 2022 Jul 12;18(7):4594. doi: 10.1021/acs.jctc.2c00500. PMID: 28430426; PMCID: PMC5717763.
- Weiner, P.K. and Kollman, P.A. (1981), AMBER: Assisted model building with energy refinement. A general program for modeling molecules and their interactions. J. Comput. Chem., 2: 287-303.
- Assisted Model Building with Energy Refinement (AMBER) - https://en.wikipedia.org/wiki/AMBER
- Jones DT, Buchan DW, Cozzetto D, Pontil M. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012 Jan 15;28(2):184-90. doi: 10.1093/bioinformatics/btr638. Epub 2011 Nov 17. PMID: 22101153.
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015 Jan;12(1):7-8. doi: 10.1038/nmeth.3213. PMID: 25549265; PMCID: PMC4428668. 10. AlphaFold decoded course by Kilian Mandon with code tutorials (GitHub)
- Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). https://doi.org/10.1038/s41586-024-07487-w
- Chai Discovery, Jacques Boitreaud, Jack Dent, Matthew McPartlon, Joshua Meier, Vinicius Reis, Alex Rogozhnikov, Kevin Wu. bioRxiv 2024.10.10.615955; doi: https://doi.org/10.1101/2024.10.10.615955
- J. Dauparas et al. ,Robust deep learning–based protein sequence design using ProteinMPNN.Science378,49-56(2022).DOI:10.1126/science.add2187
- Watson, J.L., Juergens, D., Bennett, N.R. et al. De novo design of protein structure and function with RFdiffusion. Nature 620, 1089–1100 (2023). https://doi.org/10.1038/s41586-023-06415-8
- Thomas Hayes, et. al. Simulating 500 million years of evolution with a language model, bioRxiv 2024.07.01.600583; doi: https://doi.org/10.1101/2024.07.01.600583
- Minkyung Baek et al., Accurate prediction of protein structures and interactions using a three-track neural network.Science373,871-876(2021).DOI:10.1126/science.abj8754
- AlphaFold3 release blog by DeepMind
- Chai-1 release blog by Chai labs
- ProteinMPNN blog release by Baker Lab.
- RFdiffusion blog release by Baker Lab.
- ESM3 blog release by Evolutionary Scale Lab.